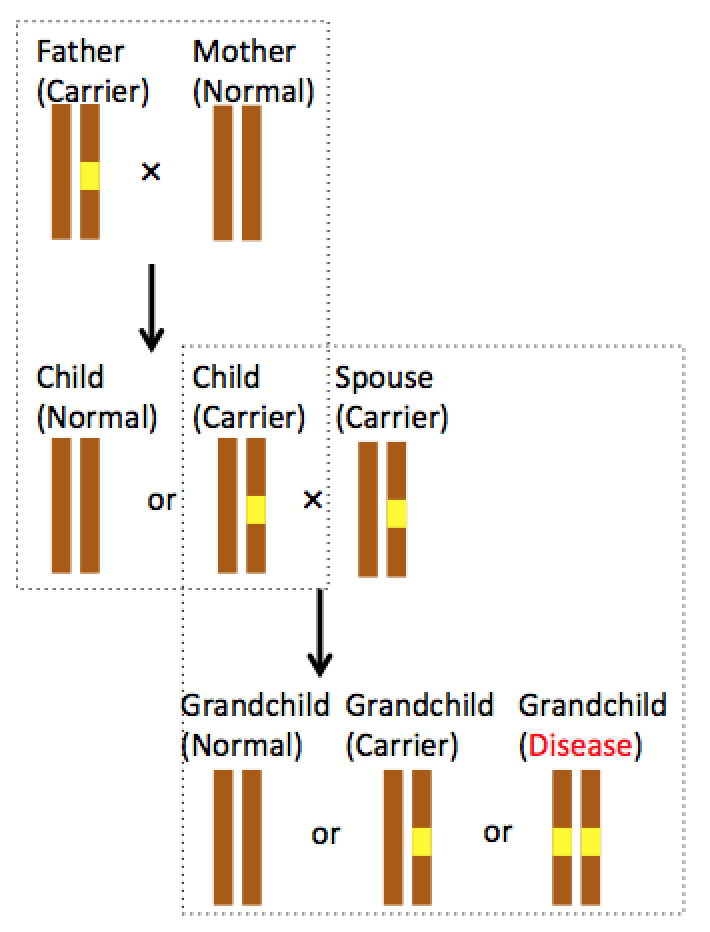
Figure 1 Disease related SNP
Figure 1 illustrates an example of how a recessive SNP cause disease. Assuming there are two different alleles in population for a given gene, one is not disease related (brown allele) and the other is disease related (yellow mixed allele). The disease related allele leads to disease in a recessive manner, meaning disease related allele produce its effect only when the gene is homozygous for disease related allele. Father carries one of each allele (heterozygosity) while mother carries only one allele (homozygosity). By Mendelian inheritance, each of their children has 50% of chance to carry the disease related allele. If the carrier child and another disease allele carrier have children, there are 25% of chance for their kids to be normal (homozygous for normal allele), 50% of chance to be carrier (heterozygous for the given gene) and 25% of chance to have the disease (homozygous for disease allele). If this disease has environmental risk factor(s), realizing one’s genome is prone to certain disease will be beneficial to avoid risk factor exposure and prevent disease from taking place, or lead to early preventative treatment.
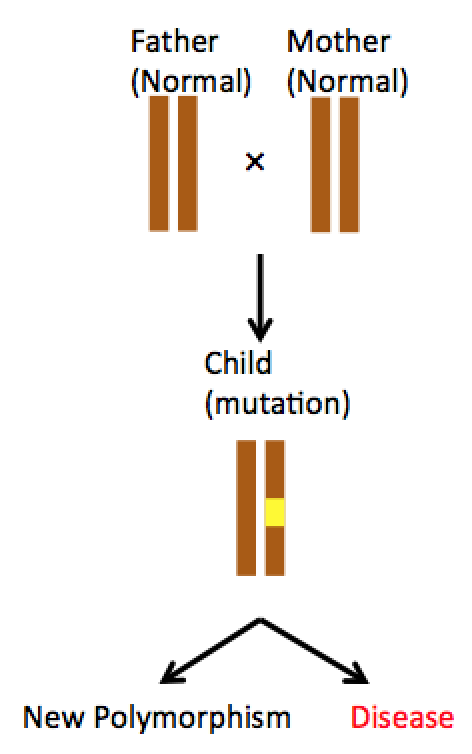
Figure 2 Mutation
Figure 2 illustrates an example of how a mutation cause disease. Mutations are not inherited from parents. Instead, mutation takes place in one’s own genome by different mechanisms.The bright side of a mutation is if it doesn’t have deteriorative effect, it can be passed onto next generation like a normal gene and circulate in population as a new polymorphism. On the other hand, not only detrimental mutations have damaged function, but also mutant’s gene product can inhibit normal allele’s product (i.e. tumor suppressor p53). In cancer research, Loss-of-heterozygosity (LOH) serves as a marker that indicates disease progression. Mutation originally took place in one of the two alleles, with the other normal allele can still produce normal gene products. When LOH is observed, both alleles are mutated, therefore the cell cannot produce normal gene products anymore.
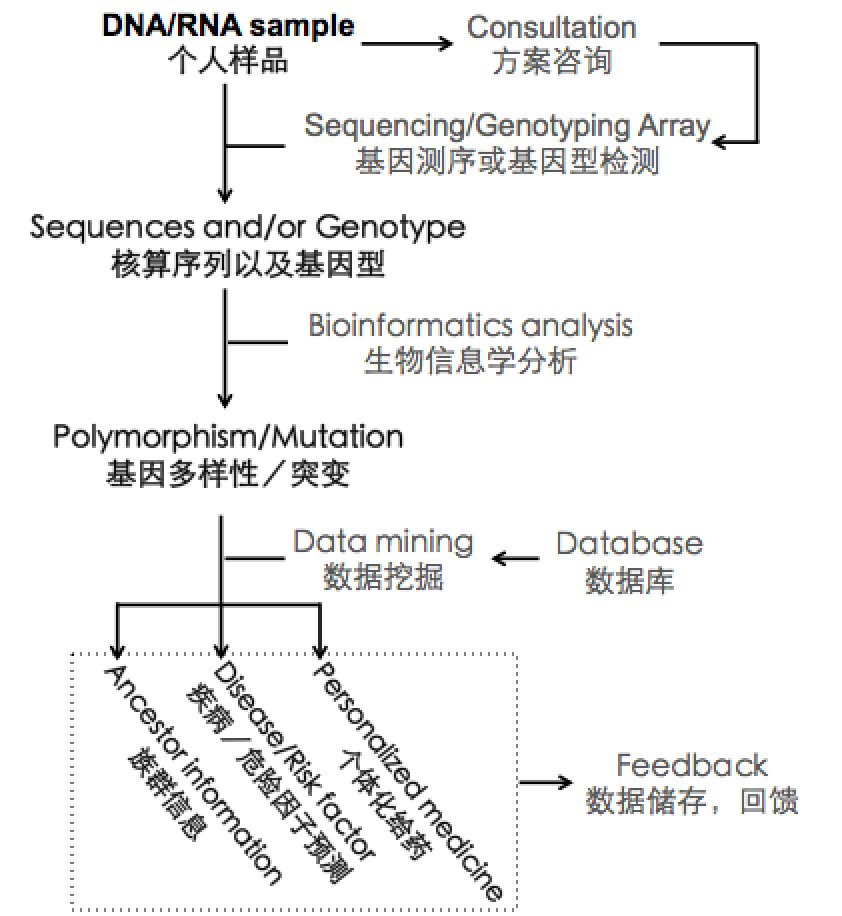
How to have personalized genome sequenced
If you are interested in sequencing your own genome, we recommend you initiate the whole process by contacting us. Based on your need, we can help you to design the best project protocol, arrange your sample collection and sequencing service. For example, if you are interested in where did you ancestor come from, we will recommend performing genotyping array plus population genetics analysis to give you the desired answer with low budget. In contrast, if you have the desire of fully sequencing your personal genome, a more complicated sequencing would be more appropriate. We will arrange your sample to be sequenced at our collaborating sequencing service provider that is close to you. After sequencing is accomplished, the sequencing data will be piped to our server for downstream analysis. Using state-of-art bioinformatics tools, we will determine gene polymorphisms and gene mutations. Subsequently, we will compare your genome to known genomic information to determine whether your genetic variation is associated with certain health condition. In the final report that will be send back to you, we will report any polymorphism and mutation we found in your genome, as well as any disease and risk factors that are potentially associated with them. We will also report any drug shown to repress the function of certain genetic variations. After the whole process, we will contact you again to go through details of your analysis result, and whether you want to share your genetic information with your health provider when necessary. If you are interested in a genome project about yourself, please contact with us.
如何检测个体化基因组
如果您希望检测自己的基因组序列,我们建议您先与我们联系。基于您的预期,我们可以帮助您设计最合适的项目流程,安排您的样品采集和测序服务。比如,如果您对您家族祖先的地理属性感兴趣,我们会推荐您做基因型分析的micro-array,以及种群遗传学的分析,而不推荐深度测序,因为前者足以满足您的需求而且价格相对便宜很多。但是如果您希望了解自己的基因组的每一个碱基序列以及所有的基因多样性,那么我们会推荐您做深度测序。在这之后,我们会安排您的基因组样品在就近的合作测序中心完成。测序结果会传送到我们的服务器上进行下游分析。我们会用最新的生物信息学工具来检测您基因组中的基因多样性和基因突变。接下来我们会将您基因组中发现的任何差异与已知的差异做比较来确定您基因组中的差异是否与特定的健康状态有关。在最终交给您的报告中,我们会列举所有检测到的基因多样性基因突变,以及这些基因组差异相关的疾病以及危险因子,同时我们也会报告有对抗致病突变活性的药物。在整个测序和分析过程完成之后,我们会再次与您联系。一方面我们会跟您解释所有的分析结果;另一方面,我们需要与您讨论事后需要将您的遗传信息分享给您的医生以方便更好的健康服务和监控。如果您对个体化基因组测序感兴趣,欢迎您与我们联系。