
Pre-experiment service
- Experiment design
- Cost estimation
- Bioinformatics consultance
- Arrange sample submission

On-experiment service
- Order sequencing service
- Arrange sequencing analysis
- Progress report

Post-experiment service/Data analysis
- Quality control and filter
- Sequence assembly
- Expression analysis
- Variance analysis
- Genome browser
- Data storage
- Data mining & Machine learning
- Publication support
NEXT GENERATION SEQUENCING
The Next Generation Sequencing (NGS) technique produces millions of short sequencing reads that represent many unconnected small pieces of a genome. With these short sequences, one may de novo construct transcripts or genomes, characterize sequence variation (i.e., single nucleotide variation (SNV), insertion, and deletion), and quantify sequence architecture (i.e., sequence repeats, copy numbers, and gene expression). Over the past decade, the sequence length of NGS (specifically Illumina technology) has significantly increased from 35 bp to current commonly produced 125 bp, and new long single sequence technology platforms (Third Generation Sequencing) are delivering sequence lengths of up to 40 kb in size (e.g., Pacific Bioscience RSII system and Sequel system) that are changing the paradigm for whole genome de novo assembly.
The Illumina genome analyzer platform is currently the most widely used NGS system accounting for over 70% of the NGS market. The sequencing process starts with preparation of a sequencing library. The DNA (for genomic sequencing) or cDNA (for RNA sequencing) sample is sheared, usually by physical, enzymatic, or chemical method, into short fragments predetermined to be a specific size, and then sequencing adaptors are ligated to both ends of each short fragment by annealing (See Figure 1). The fragments are subsequently loaded onto a flow cell, to which surface has oligonucleotides that are complementary to the adaptors bound, such that the free end of the fragment is attached to the flow cell via base pairing. A PCR step converts the initial fragment to its complementary sequence, and now both the forward strand and the reverse strand of fragments are bound to the flow cell (Figure 1). To amplify the signal, PCR is repeated for several rounds resulting in a cluster of copies around the initial fragment. Cyclic sequencing of these fragment clusters utilizes a sequence-by-synthesis process and is very similar to Sanger sequencing. One of two unique primers is attached to the free end of the bound fragments, and then nucleotides that each carries a different fluorescent reporter tag and a reversible terminator are flowed onto the flow cell. Since each nucleotide contains an elongation terminator, only a single nucleotide can be incorporated into newly synthesized sequences per sequencing cycle. After the nucleotide incorporation, laser sources excite the fluorescent reporter, and an optical sensor scans the entire flow cell to capture colors that represent newly added bases in every cluster. This optical information is converted to a base call for each growing sequence. At the end of each cycle, the terminator is removed and the next cycle continues until the desired sequence length is attained. In paired-end sequencing, after the forward strand sequence is attained, another sequencing primer initiates the sequencing of the reverse strand of each fragment.
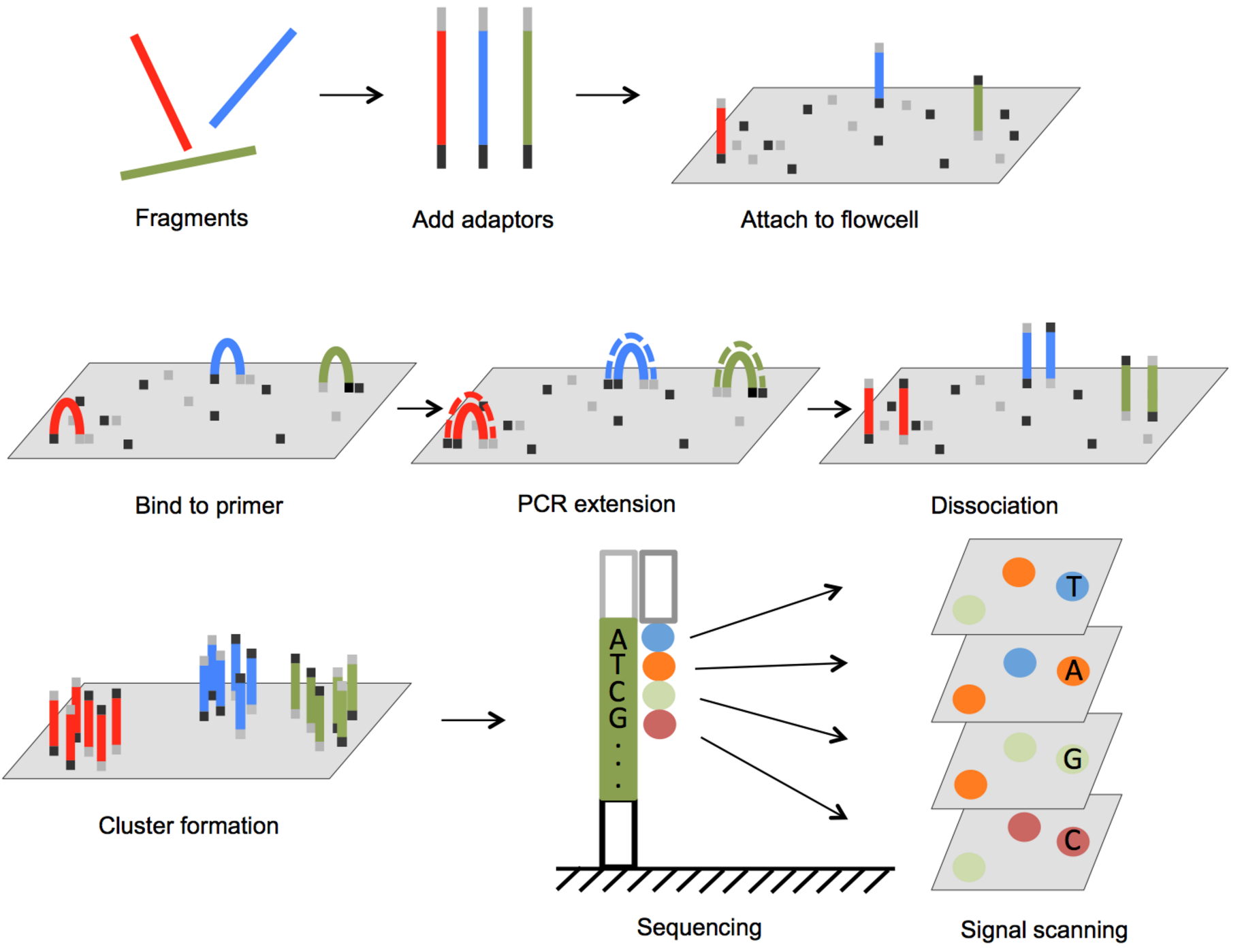
FIGURE 1 Illumina Sequencing Platform Outline of Illumina genome analyzer sequencing process. (1) Adaptors are annealed to the ends of sequence fragments. (2) Fragments bind to primer-loaded flow cell and bridge PCR reactions amplify each bound fragment to produce clusters of fragments. (3) During each sequencing cycle, one fluorophore attached nucleotide is added to the growing strands. Laser excites the fluorophores in all the fragments that are being sequenced and an optic scanner collects the signals from each fragment cluster. Then the sequencing terminator is removed and the next sequencing cycle starts.
This massively parallel sequencing platform allows high throughput sequencing. Each flow cell contains multiple lanes with each lane producing 250 million reads (i.e., up to 500 GB/flow cell) with length of each sequence read ranging from 35 bp to 250 (Illumina HiSeq-2500) or 300 bp (Illumina MiSeq). Each sequencing adaptor has incorporated into it a unique barcode in the format of oligonucleotides. Therefore multiple samples from different sources can be pooled together in one lane, and this greatly facilitates the sequencing throughput.
Before subsequent tasks such as sequence assembly or reference sequence alignment, a quality control step is usually necessary to eliminate sequencing artifacts and errors thus attain sequences that best represent the biology being studied. A short sequencing result file contains two types of “contaminants” that can hinder the sequence assembly and result in misrepresentation of actual nucleotide sequence: adaptor sequence and low quality base calls. For paired-end sequencing, the length of DNA fragment between the two adaptor sequences is defined as “insertion size.” When the desired sequencing length is longer than insertion size, the short sequencing can contain adaptor sequence in it. This artificial sequence must be trimmed off, so as not to produce significant sequence error in sequence assemblies. Another contaminant, the low quality base call, has many sources, from equipment to sequencing glitches. The quality of a base call is defined as Phred quality score (QPhred score). If we assign P as base calling error probabilities, then
Qphred=-10log10P
To retain the most usable as high-quality sequencing reads, the adaptor sequences are first clipped off, subsequently trim off low-quality base calls at the end of sequencing reads, and finally filter out sequence reads that contain a certain percentage of base calls that are below a defined QPhred score.
Pacific Bioscience offers Single Molecule Real-Time (SMRT) sequencing. In contrast to short sequencing reads provided by Illumina sequencing platforms. SMRT sequencing technology produces sequencing read length with average length over 14 kb and longest sequencing read over 40 kb (P6-C4 chemistry). SMRT sequencing technology is built upon two key innovations: zero-mode waveguides (ZMWs) and phospholinked nucleotides. ZMWs allow light to illuminate only the bottom of a well in which a DNA polymerase and DNA template complex immobilized. The labeled nucleotides allow observation of growing DNA strand.
The sequencing library is usually non-fragmented high molecular weight-DNA without PCR amplification. Bell-shaped adaptors are used to link both ends of the DNA template to form circular DNA template, which allows multiple passes of sequencing the same template (See Figure 2). The latest SMRT cell of Sequel system has 1 million ZMWs, allowing parallel sequencing of 1 million templates. Compared to RS II system, Sequel system provides 7X more sequencing reads and overall data output.
Long sequencing reads requires less sequence assembling. They are also capable of traveling through repetitive regions of genome (i.e. tandem repeats, centromere, segment duplication) that pose problems of assembling using short sequencing reads. Therefore this technology benefits the most to tasks as genome sequencing and splicing isoform sequencing.
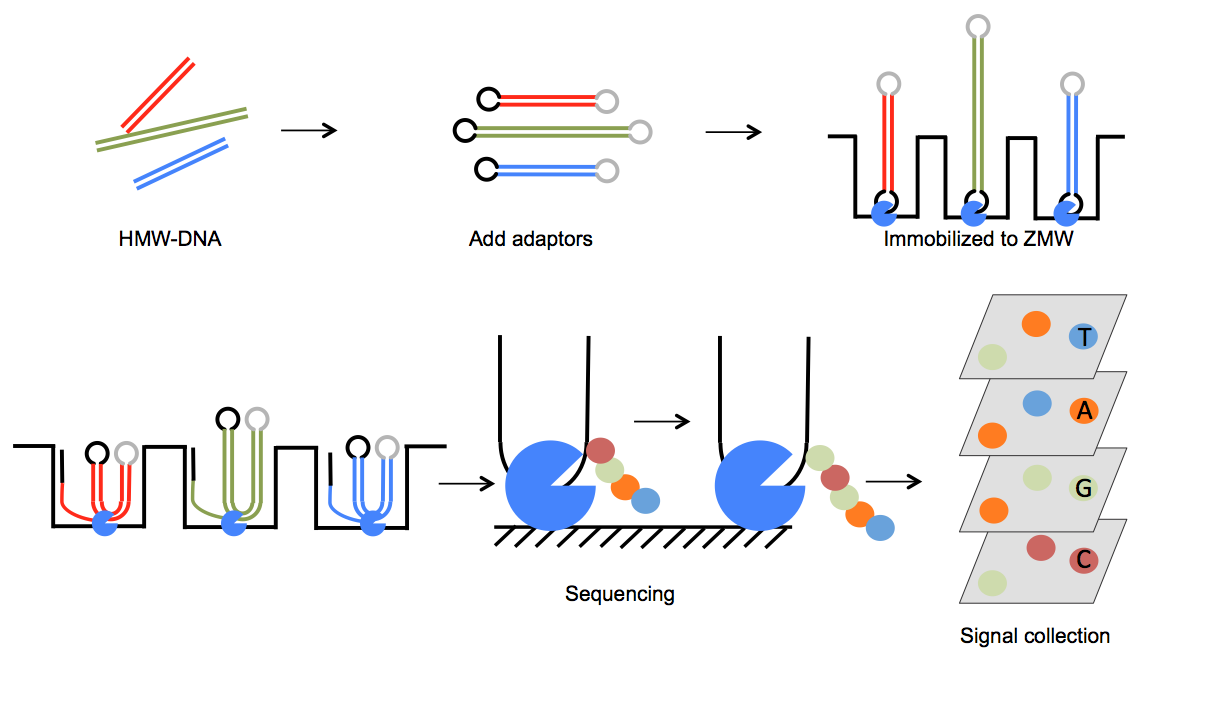
FIGURE 2 PacBio SMRT Sequencing Outline of PacBio SMRT sequencing process. High Molecular Weight-DNA (HMW-DNA) was used to create sequencing template. DNA template was immobilized to DNA polymerase attached to the bottom of zero-mode waveguides (ZMWs). Phospholinked nucleotides are applied to each ZMW chamber. Each nucleotide is labeled with a unique colored fluorophore, allowing the production and detection of unique color as new nucleotide is attached to the growing strand. Each SMRT cell contains tens of thousands of ZMWs, enable paralleled sequencing of the whole library. Sequential color information is eventually translated to DNA sequences.
For questions regarding other platforms, please contact us.
Project Design
Here in this page, options when initiating a high-throughput sequencing project are briefly introduced. These include platform selection, desired coverage or sequencing depth, insertion size, sample-pooling scheme, number of replicates, and project budget. To design the sequencing project and estimate cost, click here.
- Sequencing depth/coverage Sequencing depth or coverage reflect how many times target sequence are sequenced. These two concepts are different slightly. Coverage means how many time the genome or transcriptome is sequenced, while depth means how many sequencing read cover one nucleotide or locus. When designing a sequencing project, coverage is usually the most important factor to attain expected result in a cost-efficient way. The higher the coverage, the higher confidence the sequenced nucleotide call is real, and longer sequencing assembly, though the cost are higher. In general, genome/transcriptome assembly requires 60× to 70× coverage; Gene expression profiling requires 30× to 40× transcriptome coverage; To assess different alleles within a population, sequencing large quantity of individuals with shallow depth (i.e. 2× to 3× coverage), like what 1000 genome project has accomplished, is adequate.
- Sequencing platform Different DNA sequencing platforms produce different sequencing read lengths, sequencing quality and data output capacity, different data output format, and require different downstream data analysis pipeline. The most commonly used sequencing platforms are Hi-Seq and Mi-Seq of Illumina Inc (~70% market share). The Illumina sequencing platforms herein are referred as short sequencing technologies. They are suitable for gene expression profiling, genome/transcriptome assembly, genotyping, sequence variation discovery and .etc. Due to the limitation of short sequencing length (usually shorter than 150bp), the sequencing read, when generated from repetitive regions, cannot be explicitly assigned to a specific location of reference genome. Short sequencing reads also pose problems in assembling repetitive sequence (tandem repeats, gene duplication, centromere, segment duplication and etc.). Since the early termination of sequence assembly of assembling tool when repeats are encountered, sequence gaps (unsolved sequences) are left in assembled sequences.
Newly available sequencing technique from Pacific Biosciences (SMRT platform) offers single molecule long sequencing readings, with average sequencing length of ~14kb and maximum sequencing length over 40kb. Using PacBio sequencing platform is capable of sequencing the whole chromosome of certain species (i.e. Drosophila). Adapting PacBio sequencing platform is therefore a better choice for projects focused on sequencing assembling. Mixing Illumina with PacBio sequencing reads was also reported to generate gap-free sequence assemblies, and close existing sequence gaps within reference genome. Additionally, long sequencing technology can also provide full sequence information about transcript molecules, thus completely decipher transcript-splicing pattern. - Sample pooling scheme Illumina platforms can provide 150 million to 250 million sequencing reads per flow cell. Depends on the desired coverage, samples can be pooled together into one flow cell so that the sequencing reads are distributed into several samples. Sequencing results can be later sorted into independent files using the sample-specific barcode sequences designed in adaptor sequences.
- Sequencing length As mentioned earlier, the longer the sequencing reads, the higher chance a transcript molecule, a chromosome fragment, or even the whole chromosome can be sequenced without further sequence assembly. Therefore advantages of long sequencing reads benefit the most to sequence assembly. Not all NGS tasks require long sequencing result for optimal results. Gene expression profiling can be successfully accomplished with paired-end or single-end 50bp, 100bp or 150bp sequencing strategy. Some sequencing project (exp. Ribo-Seq), can be accomplished with 35bp sequencing reads as longer sequencing reads provides no further benefits, but only adaptor sequences.
- Library insertion size Insertion size is also referred as fragment size. Properly designed NGS project can avoid incorporating adaptor sequences into final sequencing reads, provide sequencing information from long range, estimate sequencing gap length, and bridge assembled contigs into scaffold. Having insertion size designed at 300bp and adapting 150bp paired-end read provides the same sequence information as having 300bp sequencing reads. Using mixed length of insertion size is suggested in genome sequencing project (i.e. 300bp, 800bp, 3kbp, 5kbp, 8kbp) as small library size provides seamless contig assembly and long library size scaffold contigs and estimate length of sequencing gaps. 300bp insertion size library sequenced at 60× using Illumina sequencing technology mixed with high molecular weight genomic DNA generated library sequenced at 10× depth using PacBio long-sequencing reads were proved to close sequence gaps, resolve repeat sequences and reveal new gene models in several genome sequencing project (i.e. aviation model, aquatic model).
- Number of replicates Number of biological replicates is usually a significant factor to design a successful gene expression profile project. Based on our experience, 2 biological samples from cell line samples are adequate as the variance among replicates is small. While 5-6 replicates from animal samples are required to generate statistically reliable result.
We are not a “bioinformatics” company, and we are not pursuing profit by selling products and service. We formed the group for our interest, and we want our effort to be recognized by science/research community. We do like to be on your research article if our service gives you the Eureka moment. Most of our services are free of charge. But sequencing and other experiment procedures do come with a price, high-speed computation cluster also costs money to run. In the case that requested task is computer resources heavy (de novo assembly; pileup sequencing reads; large scale data mining), we may ask you to pay for the server usage and data storage and transfer but not the bioinformatics service, unless you are collaborating with us to incorporate our service into your regular data analysis pipeline in a long-term basis.
我们并不是一个生物信息学公司,我们也不靠销售产品或服务获取利润。3402生物信息组的成立本身就是基于我们对这个领域的热情,同时我们也希望我们的努力得到领域的认可。我们的多数服务是免费的,但是测序服务和其他的实验手段,以及高性能服务器的使用都是有成本的。在用户的课题需要使用大量计算机资源的情况下(比如序列组装,测序数据与参考序列比对,大量数据挖掘),我们会要求用户支付服务器的使用,数据的保存和传输所产生的费用,而不用负担任何数据处理本身的费用,除非用户想与我们长期合作并使用我们的数据分析软件处理日常数据。